Das neue europäische Medizinprodukterecht
Allgemeines und Fristen
Lesedauer: 4 Minuten
Inhaltsverzeichnis
Mit 25. Mai 2017 sind die beiden Verordnungen
(EU) 2017/745 über Medizinprodukte (MDR) und
(EU) 2017/746 über In-vitro-Diagnostika (IVDR)
in Kraft getreten.
Im Gegensatz zu den bisherigen Richtlinien sind die beiden Verordnungen direkt anwendbar und müssen nicht in das nationale Recht umgesetzt werden.
Die MDR wird die Richtlinie 93/42/EWG über Medizinprodukte (MDD) und die Richtlinie 90/385/EWG über aktive implantierbare Medizinprodukte (AIMDD) ersetzen. Die IVDR wird die Richtlinie 98/79/EG über In-vitro-Diagnostika (IVDD) ersetzen.
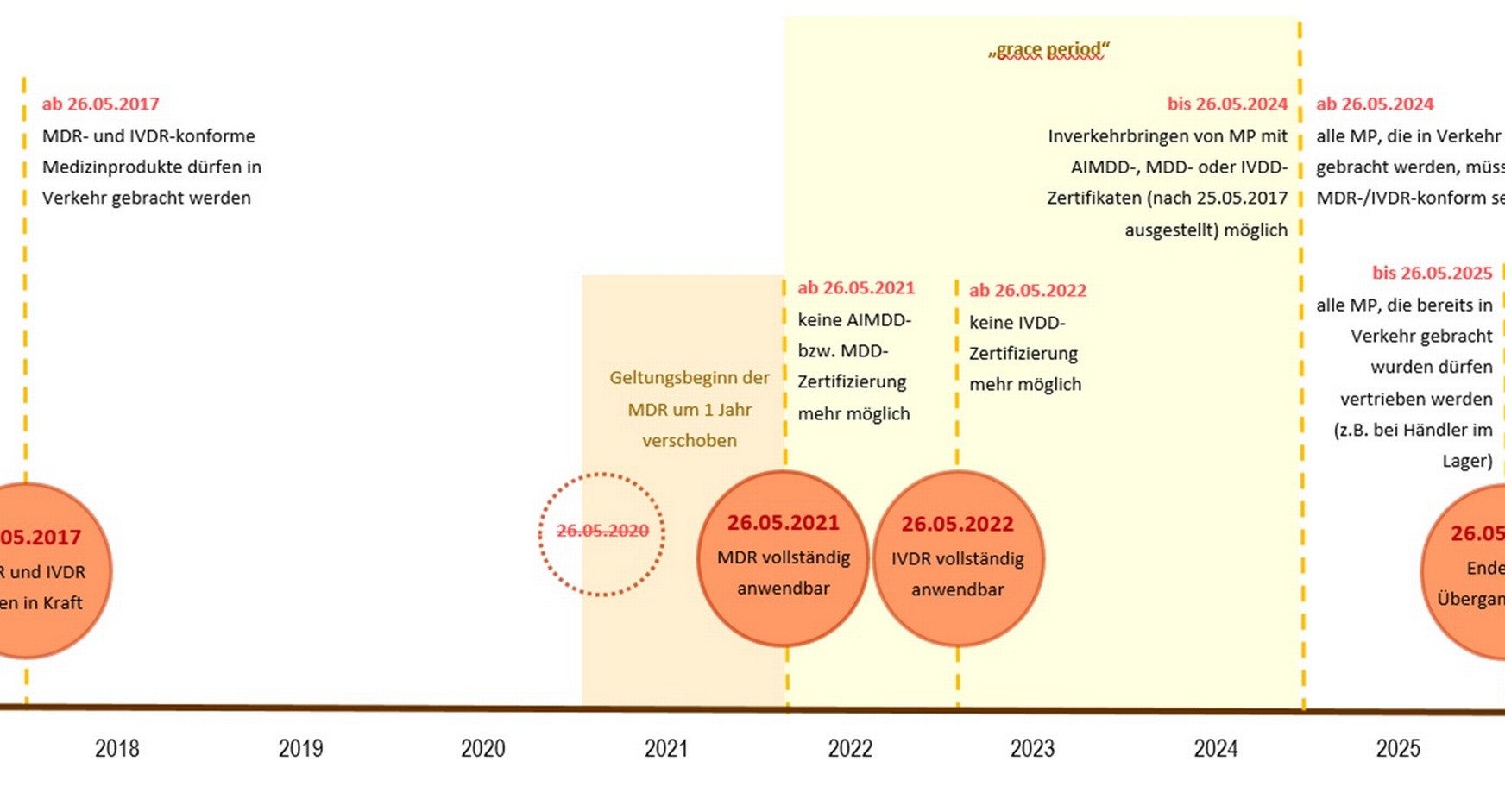
Aufgrund von Übergangsfristen sollte die Verordnung (EU) 2017/745 über Medizinprodukte (MDR) ab dem 26. Mai 2020 gelten.
Infolge der Covid-19-Pandemie wurde der Geltungsbeginn einiger Bestimmungen der Verordnung über Medizinprodukte (MDR) um ein Jahr auf den 26. Mai 2021 verschoben.
Die Verordnung (EU) 2017/746 über In-vitro-Diagnostika (IVDR) gilt ab dem 26. Mai 2022.
Mit Geltungsbeginn der Verordnungen verlieren auch die Richtlinien 93/42/EWG, 90/385/EWG und 98/79/EG ihre Gültigkeit.
Von Benannten Stellen nach den Richtlinien ausgestellte Zertifikate bleiben im Allgemeinen bis zu dem im Zertifikat angegebenen Zeitpunkt, allerdings bis längstens zum 26. Mai 2024, gültig. Nach dem 27. Mai 2024 sind Zertifikate, die im Rahmen der Richtlinien ausgestellt wurden, nicht mehr gültig.
Richtlinienkonforme Produkte, die bereits in Verkehr gebracht wurden und sich noch in der Lieferkette befinden, sind noch bis zum 26. Mai 2025 marktfähig. Danach müssen sämtliche Produkte, die bis dahin den Endanwender noch nicht erreicht haben, vom Markt genommen werden.
Übergangsfristen - Timeline:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
FAQ Übergangsbestimmungen:
Die neuen EU-Verordnungen über Medizinprodukte und In-vitro Diagnostika bringen einige Neuerungen und sehen erhöhte Anforderungen für die Wirtschaftsakteure vor. So wurden in den Verordnungen die Pflichten der Hersteller, Bevollmächtigten, Importeure und Händler präzisiert. Strengere Anforderungen bei der Produktzulassung und an die Benannten Stellen, ein erweiterter Geltungsbereich und die Neuklassifizierung bestimmter Produkte soll die Sicherheit der Medizinprodukte erhöhen. Die Einführung einer einmaligen Kennnummer, der „UDI – Unique Device Identifiers“ soll die Rückverfolgbarkeit entlang der gesamten Lieferkette bis hin zum Endverbraucher oder Patienten gewährleisten. Für mehr Transparenz sorgt die erweiterte Rolle der Europäischen Datenbank für Medizinprodukte EUDAMED.
Um Marktstörungen und die Nichtverfügbarkeit von Medizinprodukten zu vermeiden sowie einen möglichst reibungslosen Übergang von den Richtlinien zu den Verordnungen zu ermöglichen, gibt es Übergangsfristen und –bestimmungen.
Ja, Zertifikate bleiben in der Regel bis zum Ende des auf dem Zertifikat angegebenen Zeitraums oder bis zum 26. Mai 2024 gültig (je nachdem welcher Zeitpunkt früher liegt). Danach sind Zertifikate, welche von benannten Stellen gemäß den Richtlinien erstellt wurden, ungültig.
Nein! Bis Mai 2025 werden nach den Richtlinien zertifizierte Produkte und Produkte, die im Rahmen der Verordnungen zertifiziert sind, nebeneinander auf dem Markt angeboten.
Beide haben nach dem Gesetz den gleichen Status, und es darf keine Diskriminierung bei öffentlichen Ausschreibungen stattfinden.
Nein, Bescheinigungen für Produkte, die nach den neuen Verordnungen in eine höhere Risikoklasse eingestuft wurden, bleiben bis zu ihrem Ablaufdatum gültig.
Ja, jenen Medizinprodukten der Klasse I, die aufgrund der MDR eine Höherklassifizierung auf Ir, IIa, IIb oder III erfahren haben und daher durch eine Benannte Stelle zertifiziert werden müssen, wurde eine zusätzliche Frist von vier Jahren eingeräumt: Die Produkte dürfen nun bis zum 26. Mai 2024 auf den Markt gebracht werden (Art 120 Abs 3 MDR).
Gemäß Art 120 Abs 4 MDR können richtlinienkonforme Medizinprodukte, die bereits in Verkehr gebracht wurden, bis zum 26. Mai 2025 am Markt bereitgestellt oder in Betrieb genommen werden - vorausgesetzt diese Produkte wurden vor dem 26. Mai 2021 in Verkehr gebracht oder fallen unter die Ausnahmeregelung des Art 120 Abs 3 MDR.
Weitere Informationen finden Sie in unserem Rechtslexikon für Medizinprodukte.
Informationsblätter der Europäischen Kommission
Die Europäische Kommission hat Factsheets zu den beiden EU-Verordnungen MDR und IVDR auf Ihrer Seite herausgegeben: https://ec.europa.eu/health/md_newregulations/publications_de
Darin enthalten sind grundlegende Informationen zu den Neuerungen, Änderungen, Geltungsbeginn, Übergangszeiten sowie häufig gestellte Fragen im Zusammenhang mit den neuen Verordnungen.
Auf der Website der Europäischen Kommission sind auch die Leitfäden der Koordinierungsgruppe für Medizinprodukte veröffentlicht. Diese dienen zur Sicherstellung einer einheitlichen Durchführung der Verordnungen innerhalb der EU: https://ec.europa.eu/health/md_newregulations/guidance_en